Article written by Vera Siffnerova (FAS, Omixon)
In most Slavic languages, LOH stands for Summer Olympic Games (e. g. Letní Olympijské Hry in Czech), in HLA world, LOH is known as an abbreviation of an important disease marker – Loss of Heterozygosity – during which different types of cancer cells try to escape immunity mechanisms to win their own “survival game“.
Looking at this phenomenon from a wider perspective, loss or down-regulation of HLA Class I antigens in tumor cells has been frequently observed in a variety of human malignancies(1-5). The first description of MHC Class I loss was done in a mouse model (Gardener lymphoma) by Dr. Festenstein in 1976(6). This mechanism is not an exclusivity of cancer cells, but also of viruses. It has been found that viruses use a similar mechanism to avoid recognition and elimination by the immune system(7).
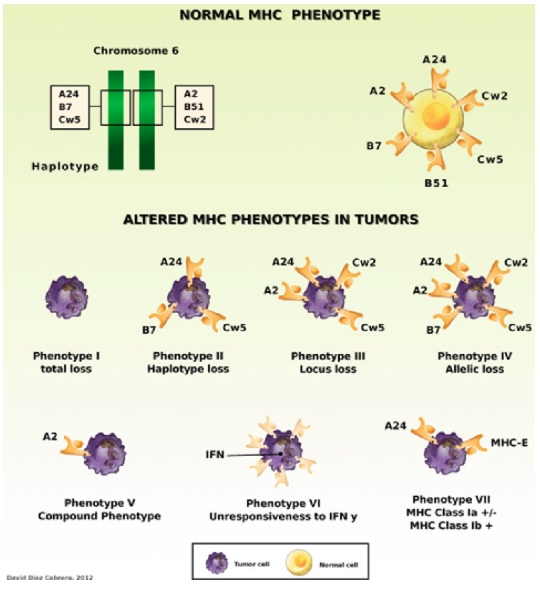
Figure 1. Examples of altered MHC class I phenotypes found in human tumors. HLA Class I molecules can be totally or partially absent from tumor cells (Phenotype I to Phenotype V). In some cases, tumor cells do not change Class I expression after treatment with IFN-γ (Phenotype VI). Some tumor cells express aberrant HLA-E molecules together with low expression of HLA-A, -B, or -C classical class I antigens (Phenotype VII)(8).
Recent advances in HLA haploidentical hematopoietic stem cell transplantation (haplo-HSCT) have been associated with significant reduction in treatment-related mortality. However, the most common cause of treatment failure, as with any other type of allogeneic transplant, remains to be the disease relapse. While, in general, etiology of relapses after haploidentical transplantation may overlap with other types of allogeneic transplants, a unique relapse mechanism with loss of heterozygosity is only observed after haplo-identical transplantation. This kind of transplantations may offer augmented graft-versus-leukemia (GvL) effect owing to its unique treatment platform, HLA disparity and, in certain situations, enhanced natural killer (NK) cell alloreactivity. Preemptive infusions of modified donor lymphocytes, NK cells, or chimeric antigen receptor (CAR) T-cells after haploidentical transplantation are interesting areas of ongoing research to prevent and treat relapses(9).
LOH as a possible mechanism of leukemia immune escape after haplo-HCT has been described by several groups. Vago and coworkers have studied the genomic rearrangements in mutant variants of leukemia by using genomic HLA typing, microsatellite mapping, and single nucleotide polymorphism (SNP) arrays. They identified the specific mutant variant of leukemia cells with HLA that differed from the donor’s haplotype which had been lost because of acquired uniparental disomy (aUPD) of chromosome 6p in 5 of 17 patients with leukemia relapse after haploidentical transplantation. This mutation resulted in leukemic cell evasion from donor T-cell recognition, whereas the original leukemic cells taken at the time of diagnosis were efficiently recognized and killed. The authors hypothesized that HLA loss may reflect alloimmune pressure mediated by donor T-cells toward the HLA mismatches(10).
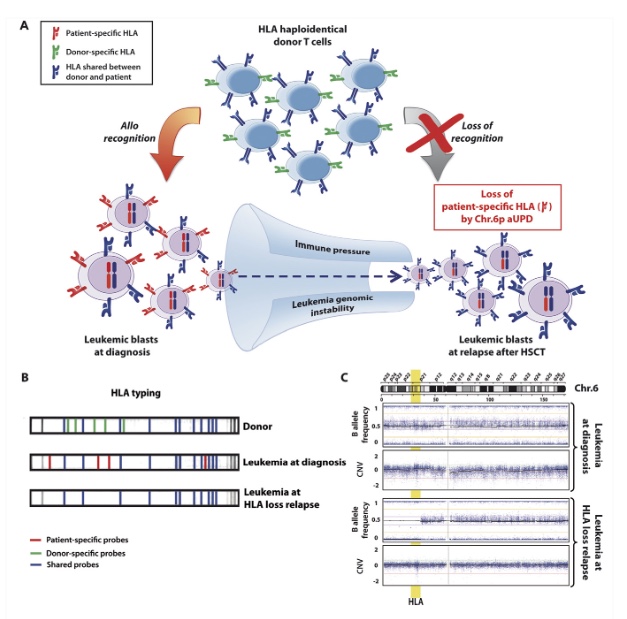
Figure 2. Loss of mismatched HLA in leukemic cells after haploidentical HSCT. (A) Biological basis and consequences of genomic loss of the patient-specific HLA haplotype after transplantation. Leukemic cells, heterozygous at diagnosis (on the left), are subject to an intense immunologic stress after transplantation, mostly mediated by donor T cells targeting the mismatched HLA haplotype (in red). This pressure, combined with the genomic instability intrinsic to tumor cells, leads to the generation and selection of mutant variants that lack the patient-specific HLA haplotype (on the right) and therefore are no longer recognized by donor T lymphocytes. (B) Schematic representation of low-resolution HLA typing by forward PCR sequence-specific oligonucleotide probing (SSOP) of DNA from a donor and a haploidentical patient’s leukemia at diagnosis or at HLA loss relapse after transplantation. The lines on each strip represent signals of probes specifically hybridizing to the HLA alleles amplified from the DNA under analysis. Signals for HLA alleles from the shared haplotypes are shown in black, signals for HLA alleles from the patient- and the donor-specific haplotype are shown in red and blue, respectively. Note the absence of patient-specific signals in the purified leukemic blasts at relapse, demonstrating genomic loss of the corresponding allele. (C) SNP profile of chromosome 6 of purified AML blasts harvested at diagnosis (upper dot plots) and at relapse after haploidentical HSCT (lower dot plots) (adapted from Toffalori et al46). Upper plots show the B allele frequency, which indicates the zygosity of each SNP: a physiological situation that comprises equal representation of AA, AB, and BB genotypes, respectively, displaying B allele frequencies of 0, 0.5, and 1, as observed in blasts at diagnosis. Lower plots show the LogR ratio, which is a measure of SNP CNV, with a normal value defined as 0, as at diagnosis. Note in AML at relapse the large region of the short arm of chromosome 6 without heterozygous SNPs (absence of dots with B allele frequency 0.5) but with conserved CNV, demonstrating de novo aUPD encompassing the MHC (in yellow)(11).
This phenomenon was also confirmed in a report by Villalobos and coworkers, describing two cases of HLA loss of chromosome 6p aUPD that resulted in a total loss of the mismatched HLA haplotype among three pediatric patients with AML who relapsed after haplo-HSCT. The molecular events that form the basis of this type of genomic abnormality remain uncertain, but it has been postulated that aUPD may derive either from mitotic homologous recombination events or from an attempt to correct for the unbalanced loss of chromosomal material by using the remaining alleles as a template resulting in copy number neutral-LOH without a concurrent change in the copy number; therefore, standard cytogenetic methods fail to detect this phenomenon(12).
Another mechanism of LOH has been described by McCurdy et al in a study of two high-risk AML patients who relapsed after haplo-HCT using posttransplant cyclophosphamide. In this report, the authors demonstrated the absence of a mismatched recipient HLA haplotype on the isolated leukemic blasts in both cases. Interestingly, both cases represent distinct mechanisms of HLA loss. SNP array for recipient 1 demonstrated aUPD at 6p, which is the same mechanism that was previously described. However, the karyotype and SNP array for recipient 2 revealed a deletion of chromosome 6p that encompassed the mismatched HLA locus. The later represents a different, but similar, genomic mechanism and supports that the leukemic cells may lose the mismatched HLA haplotype through multiple means, resulting in evasion of the donor immune system(13).
The last example of mechanism causing down-regulation of mismatched HLA class I antigens was described by Tamaki et al. In this study, this group found a lack of mismatched HLA-A using flow cytometric analysis, despite the retention of both HLA haplotypes on the leukemic cell surface of the AML patient who relapsed after haplo-HSCT. They speculated that this finding might be associated with impaired epigenetic regulation of the gene causing down-regulation of HLA class I on unshared alleles, which are preserved on shared alleles(14).
It becomes crucial to document the HLA loss in patients who have leukemia relapse after haplo-HSCT because it has several relevant clinical consequences. Not only does it demonstrate that the donor-derived T-cells circulating in the patient at the time of relapse become inefficient bystanders, but also that any attempt to induce remission by infusion of donor T-lymphocytes is expected to be ineffective against the leukemic cells and potentially harmful to the patient due to the conserved risk of inducing GvHD(15).
Based on the net result of the genomic alteration, two possible alternative immunotherapeutic strategies can be considered for these variants of leukemia relapse. The first is a second transplantation from a different HLA-haploidentical donor, selected for being mismatched against the HLA haplotype retained by leukemic blasts. Theoretically, the advantage of this method is that the donor and leukemia cells would have a full immunologic incompatibility that can help increase GvL effect, while an incompatibility between the donor and patient’s healthy tissues is only 50%. The second treatment option is an infusion of high-dose purified donor NK cells. It is based on an observation that leukemic cells that undergo genomic loss of one HLA haplotype in several cases also lose the ligands for donor inhibitory KIRs, becoming in principle susceptible to NK cell alloreactivity. Even though, the effectiveness of this strategy is limited in overt leukemia relapse, it might help in preemptive treatment of impending leukemia recurrence, guided by molecular markers of minimal residual disease and early detection of HLA loss relapse(16).
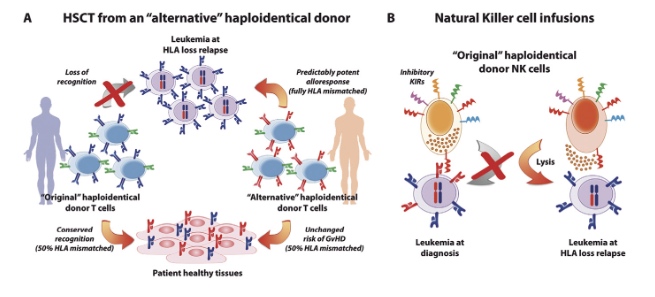
Figure 3. Immunotherapeutic strategies for the treatment of HLA loss relapses. (A) Second transplantation from an “alternative” haploidentical donor. T cells from the “original” haploidentical donor (on the left) have lost their GvL potential because they are no longer alloreactive to leukemic cells carrying genomic loss of the mismatched HLA (in red), while they conserve their potential to cause GvHD by attacking the patient’s healthy tissues. Conversely, T cells from an “alternative” haploidentical donor (on the right), matched with the patient for the HLA haplotype lost by leukemia (in red), are predictably alloreactive to the mutant leukemia, compared to which they are fully HLA mismatched, while displaying a risk of eliciting GvHD comparable to that of T cells from the “original” donor. (B) NK cell infusions. Donor NK cells express a wide set of inhibitory KIRs, among which those recognizing as their ligands the patient-specific class I HLA molecules (in red) may impair NK cell-mediated GvL against leukemia at diagnosis (on the left). Loss of the patient-specific HLA at post-transplantation relapse may result in abolishing this interaction, and enable donor NK cells to eliminate the mutated leukemia (on the right). This could be exploited by infusing freshly harvested or ex vivo activated NK cells from the “original” donor(11).
Haplo-HSCT has evolved from a high-risk procedure to a widely acceptable alternative donor transplant option with similar toxicity and outcomes compared to HLA-matched donor transplants in retrospective studies. Loss of heterozygosity aspects and related studies results have been successfully used to improve transplantation outcomes. Ongoing continuous investigations on this topic still bring more questions and some uncertainties (similar to any other research). Once answered or understood, LOH affairs can possibly be nominated as a new top sport at HLA World Olympic transplant playground.
References:
- Garrido F, at al (1997) HLA and cancer: 12th international histocompatibility workshop study. HLA, Genetic diversity of HLA. Functional and medical implications. vol. I. In: Charron D, EDK (eds). pp 445–452
- Marincola FM, et al (2000) Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74: 181–273
- Campoli M, et al (2002) HLA class I antigen loss, tumor immune escape and immune selection. Vaccine 20(Suppl 4):A40–A45
- Chang CC, et al (2005) Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res 93:189–234
- Aptsiauri N, et al (2007) Role of altered expression of HLA class I molecules in cancer progression. Adv Exp Med Biol 601:123–131
- Garrido F, et al (1976) Further evidence for depression of H-2 and Ia-like-specificities of foreign haplotypes in mouse tumour cell lines. Nature 261(5562):705–707
- Ploegh HL. (1998) Viral strategies of immune evasion. Science 280(5361): 248–253.
- Aptsiauri N,et al (2013) MHC Class I antigens in malignant cells: immune escape and response to immunotherapy. New York: Springer Briefs in Cancer Research
- Gaballa S, et al. (2018). Prevention and Treatment of Relapse After HLA-Haploidentical Hematopoietic Cell Transplantation. 10.1007/978-3-319-54310-9_19.
- Vago L, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361(5):478–88.
- Vago L, et al. Genomic loss of mismatched human leukocyte antigen and leukemia immune escape from haploidentical graft-versus-leukemia. Seminars in oncology 39 6 (2012): 707-15 .
- Villalobos IB, et al. Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood. 2010;115(15):3158–61.
- McCurdy SR, et all. Loss of the mismatched human leukocyte antigen haplotype in two acute myelogenous leukemia relapses after haploidentical bone marrow transplantation with post-transplantation cyclophosphamide. Leukemia. 2016;30(10):2102–6.
- Tamaki H, et al. Different mechanisms causing loss of mismatched human leukocyte antigens in relapsing t(6;11)(q27;q23) acute myeloid leukemia after haploidentical transplantation. Eur J Haematol. 2012;89(6):497–500.
- Barrett J, Blazar BR. Genetic trickery-escape of leukemia from immune attack. N Engl J Med. 2009;361(5):524–5.
- Einsele H., et al (2018) Unique Complications and Limitations of Haploidentical Hematopoietic Cell Transplant. In: Ciurea S., Handgretinger R. (eds) Haploidentical Transplantation. Advances and Controversies in Hematopoietic Transplantation and Cell Therapy. Springer, Cham