What is non-targeted data?
For the sake of this post, ‘non-targeted data’ will be used as a collective term for data with more than a dozen or so targeted genes. Whole exome or whole genome sequencing, whole MHC, or multi-gene panels all fall into this category. These data types can (of course) be very different from each other and significant differences within the same category are also expected depending on the exact experiment design. What sequencing data generated from these sequencing methods share is that they have very different characteristics compared to targeted data.
Targeting and HLA
There are countless methods for generating non-targeted data. Enrichment is usually done with either PCR or PCR-free methods. The problem is, that methods not specifically aimed at the MHC region often have trouble with picking up the extremely diverse HLA genes. There are many assays that only use the relevant region of chromosome 6 and the known alternative haplotype sequences available as part of the human reference genome as a basis for targeting design and some enrichment methods skip most of the MHC region entirely. So, before spending a huge amount of money on sequencing always make sure that your precious data will actually contain the genes you are interested in.
Note, that missing some (or even most) HLA alleles with generic enrichment methods is not a fault and gives basically no information about the ‘goodness’ of the method. Specialised HLA genotyping methods exist for a reason. Designing primers for even a single HLA locus can be challenging (more than 3000 alleles are defined for HLA-A only(!)). So in general, be prepared for seeing the lower end of the expected coverage for HLA genes in non-targeted data.
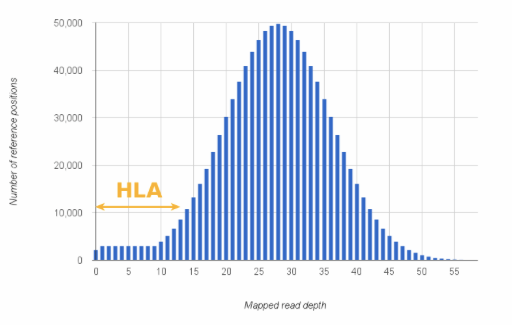
Figure 1: Coverage depth distribution of a hypothetical whole exome sequencing experiment.
Sources for non-targeted data
If you don’t have pots of money laying around that you can freely use for sequencing or you just want to play around a little, you have quite a few options for getting non-targeted data. More than 3000 thousand samples are freely downloadable from the 1000 Genomes Projects website and most of these were sequenced with multiple methods. If you are looking for something more exotic, it’s always a safe bet to check one of the short read archives. If you are interested in high quality and high coverage Illumina data, you can take a look at the Illumina Platinum Genome set. There are also other interesting ongoing projects which might worth a look, like the 100,000 Genomes Project. You can find a long list of these on the following site, collected by the creators of Kaviar.
Data formats
For many samples, it is possible to get raw sequencing data (i.e. either fastq files, or something else, that can be converted back to fastq, e.g. SRA). On the other hand, raw data cannot be directly accessed for some samples, but alignments can still be downloaded. These alignments are usually in either the classic BAM format or something more sophisticated like CRAM. If you want to use alignments as an input for HLA genotyping, make sure that the files contain unmapped reads, as it is very likely that a significant proportion of reads originating from HLA genes will end up in this category.
HLA genotyping from non-targeted data
As file sizes for non-targeted data can be huge, especially if alignment files are used as an input (see Figure 2), adding a filtering step to the HLA genotyping pipeline usually makes sense. The best way to do this is to align all the reads to the whole IMGT/HLA database and collect reads matching any of the alleles to a new file. Note, that due to the high level of partially defined alleles in the database, generic short read aligners designed with long, continuous reference sequences in mind might not be suitable for the job. The filtering step also makes sense, because the resulting smaller files are much more suitable for re-analysis with different methods or parameter sets.
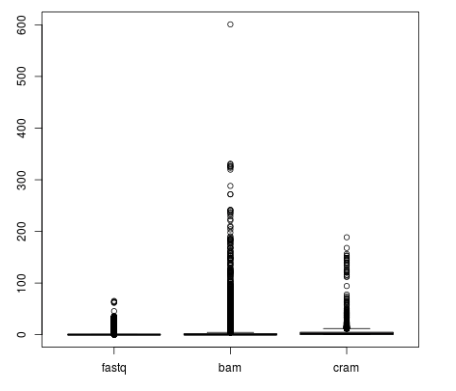
Figure 2: File sizes in GB from the 1000 Genomes Project grouped by file type.
After filtering, genotyping can be attempted. Depending on data quality and coverage patterns, different methods can be tried. For high coverage data where full HLA genes are covered, assembly based methods can be used. For whole exome and genome data, alignment based genotyping methods are more suitable due to the low and/or uneven coverage and coverage depth.
How do the results look like?
For whole genome and whole exome data, the following characteristics will likely be observed for at least some of the loci:
- highly ambiguous genotypes,
- dodgy coverage,
- full coverage gaps,
- dropouts (i.e. loci where no alleles were called),
- incorrect genotypes.
Why use non-targeted strategy at all?
If the data is (at least potentially) so bad, why not use targeted data instead? There are many possible reasons. For example:
- You are looking for causative genes/variants, but have no idea where to look (e.g. rare diseases).
- Several loci are of interest.
- You don’t have/don’t want to design primers for a hundred genes.
- You won a grant for WGS/WXS.
- You might want to use the data for something else later.
- …
Let’s take a look at a few samples!
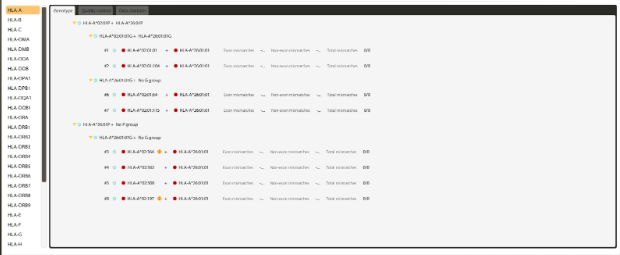
Figure 3: Highly ambiguous HLA-A result from a whole exome sequencing (WES) experiment.
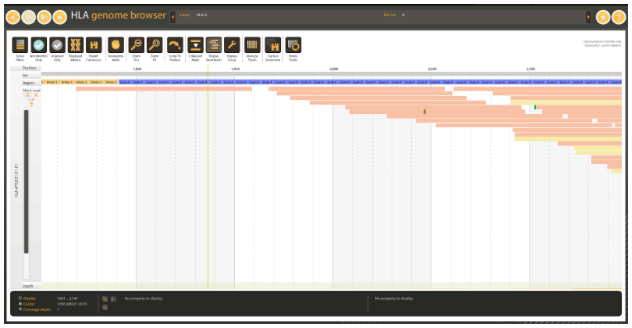
Figure 4: Exonic region covered by a single read in WES data.
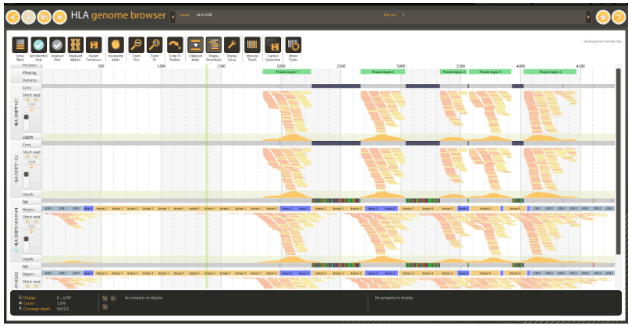
Figure 5: Consensus contigs generated from high coverage whole exome data. Note the uncovered regions and corresponding consensus gaps.

Figure 6: HLA alignment in a randomly selected sample from the 1000 Genomes Project. Note, that this sample does not have an unusually low quality (at least outside the MHC region).
What (else) can be done with non-targeted data?
As you could see on the previous figures, using non-targeted (especially low coverage whole genome) data comes with a whole box of problems when it comes to HLA genotyping. Fortunately, there are many paths that can be taken which can make all the trouble (and money) worthwhile. Just to give you a few examples:
- association studies (beware of LD, including non-HLA genes),
- causative gene/variant hunting,
- getting sequences for not yet documented alleles,
- haplotyping (with family data),
- population genetics,
- phylogenetics,
- gene prediction,
- homozygosity rate,
- crossover events,
- …
Take-home message
All in all, non-targeted data analysis, and more specifically HLA genotyping from this data type has its highs and lows. My suggestions would be the following:
- Don’t be hasty! – Plan your experiment and select your method very carefully.
- Try before you buy! – Try to get some representative example data or even consider a pilot project.
- It’s not one-size-fits-all! – Experiment with genotyping parameters until you find the one that suits your data.
- Double-check before going big! – Consider doing some validation to assess genotyping accuracy.
- Keep your expectations realistic!
/ Written by Krisztina Rigó