Written by Nina Lauterbach (FAS, Omixon)

About 3,500 years ago, a scholar in Egypt started writing in the oldest medical text known in history. One of the diseases he described was something called Resh. The name is weird and unfamiliar, but the symptoms might ring a bell, a cough and a flowing of mucus from the nose. He was describing the human rhino virus, or in other words, the common cold. To this day, the virus is still very much part of our lives, and it has been estimated that every person will spend a year of his or her life lying in bed, sick with colds. Even though we had all the time in the world to find a cure, it never came. We have no choice but to go through the illness many times in our lives and every time we must trust our immune system to win the battle. Luckily, the rhino virus causes little harm and even though we feel miserable for a few days, we always recover. The reason why it manages to infect us multiple times across our lifespan, and why we have not developed a cure yet, lies in the genetic diversity of this virus. There is a certain part of the virus which has mutated over the course of time creating many different strains. It is because of these numerous strains that a virus can avoid being killed by our immune system and is able to spread to another host, who might be your partner or colleague. The rhino virus is not very harmful to us, which is great, but that is actually one of the reasons why it is so successful at surviving amongst us. The better we feel, the more we interact with others and the easier it spreads to another human host. Which in the end, is the goal of every virus (1-3).
Unfortunately, it is not only the common cold virus we have to deal with. We have been sharing our World with a tremendous amount of viruses and they have been in our lives for so long that our own genome even contains viral DNA (4). In fact, if all the 1×10³¹ viruses on Earth were laid end to end, they would stretch for 100 million light years (5). Luckily, it has been estimated that “only” about 219 viruses are able to infect and compose a thread to us humans (6). To be able for us to survive amongst all these viruses we rely on our immune system to work as a barrier and defense mechanism. Too bad for us that viral pathogens have “learned” how to exploit our immune system and modulate or evade immune responses that would otherwise eradicate them (7).
So, evolution has put an immense pressure on viruses to find ways to circumvent elimination by the host’s immune response. Therefore, some viruses have created a whole array of tools to prevent from being recognized and targeted by our immune system. One of the most sophisticated and successful viruses in this respect known today is the human cytomegalovirus (HCMV). More than half of the global human population is latently infected with HCMV (8), in other words, in these people the virus lays dormant waiting to be reactivated. A healthy adult immune system is rather effective in preventing HCMV-associated diseases, however once the immune system is either immature or compromised, HCMV infections frequently progress into life-threatening diseases. Therefore, the most vulnerable people, such as transplant recipients, AIDS patients, and congenitally infected newborns, suffer the most from HCMV (9). But what makes this virus so successful at evading our well-developed defense mechanisms?
Since presentation of viral peptides by HLA class I cell surface molecules to T cells is critical in getting rid of viral infected cells, HCMV releases a set of glycoproteins upon entering the cell that interfere with almost all stages of the HLA class I antigen presentation pathway to avoid T cell recognition. For example, glycoprotein US2 and US11 redirect the HLA class I molecule from the endoplasmic reticulum to the cytosol and will induce the degradation of the HLA class I molecule. While, glycoprotein US3 inhibits the loading of the viral peptide to the HLA class I molecule and retains it in the endoplasmic reticulum (10). One other of HCMV’s attributes is the talent of mimicking aspects of our immune system. For example, the CMV- encoded HLA class I-like protein UL18 binds to the inhibitory receptor LIR1 on NK cells to inhibit the NK cell from killing the HCMV infected cell, and it does this with even stronger affinity than HLA class I (11). Furthermore, HCMV releases the UL40 peptide which is very similar to our own HLA-E ligands, and thereby it can bind to HLA-E and maintain its expression on the cell surface, ones again to avoid NK cell killing (Figure 1). The virus has learned how to imitate elements of our immune system and even improve them, to avoid being recognized by T cells, and to engage inhibitory receptors to turn off the NK cells. Interestingly, HIV-1 and hepatitis C virus use the same mimicking strategy (12, 13).
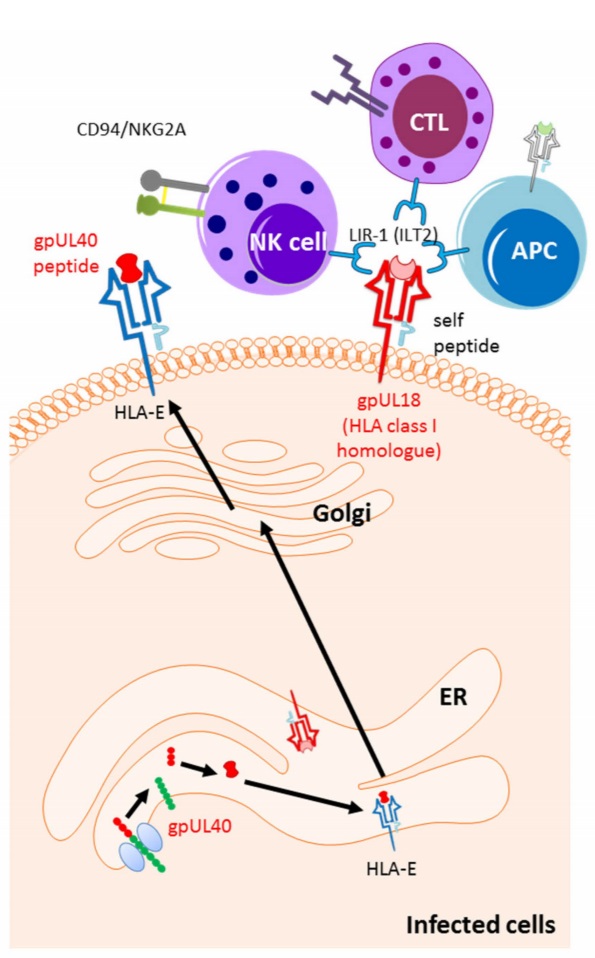
Figure 1: HCMV-mediated up-regulation of the ligands for NK cell inhibitory receptors. The signal peptide of unique long UL40 up-regulates the surface expression of HLA-E. HLA-E is a ligand for the NK cell inhibitory receptor CD94/NKG2A. The HLA class I homologue, UL18, inhibits the NK cell activity by inducing NK cell inhibitory receptor LIR1 (10).
Next to the direct effects of HCMV on viral peptide presentation, HCMV releases a homologue of the immune modulator IL-10. HCMV IL-10, same as the human IL-10, possesses potent immunosuppressive properties, such as the inhibition of proinflammatory cytokine synthesis, and downregulation of cell surface expression of both HLA class I and class II molecules (14, 15). Enhancing even more the chances of the virus to outsmart our immune surveillance. And, these are just some parts of the array of immune evasive strategies that a virus like HCMV uses. The capacity to evade host immunity is a property associated with many different viruses, especially among the herpesvirus family, such as Epstein–Barr virus, herpes simplex virus 1 and herpes simplex virus 2. Comparison of the molecules they target and strategies they use reveals many similarities to those described for HCMV.
So, HCMV is a “smart” virus that has infected many people all over the world (in some developing countries even all), however most people will never even know whether they carry the virus inside them as they will never develop the symptoms. This is different with a virus like influenza. The sentence “I caught the flu, so I’m going to stay in bed” is something that almost all people have said more than once in their life. Just like the common cold, the flu virus is rooted into our existence, however the symptoms are typically much worse. Fever, cough, sore throat, muscle aches, headaches, are just some of the “fun” trades that come with catching the flu. Luckily, most people who get the flu will recover in less than two weeks. But in some people the flu causes complications, including pneumonia and even death. Worldwide, the annual influenza epidemics result in about 3 to 5 million cases of severe illness, and about 290 000 to 650 000 respiratory deaths (16). This may sound like a high number however it is not even close to the number of people killed by flu pandemics in the past, as from 1918 until 1919 influenza was responsible for more than 50 million deaths worldwide. Fortunately, in our modern society, infectious diseases are not as deadly anymore, due to vaccine programs, antimicrobial treatments, and improved surveillance.
Due to influenza’s ability to change its genetic makeup it still allows for the disease to grow to pandemic proportion, and it makes it impossible to generate a universal vaccine. Just like the common cold virus, influenza has a high mutation rate (also seen with other notorious pathogens as HIV, SARS, MERS, Zika, Ebola and polio), which occurs through two processes called antigenic drift and antigenic shift (Figure 2). Antigenic drift occurs when mutations in the viral strains accumulate over time, which likely will not alter the virus drastically, and it will still be recognizable to antibodies in the body. However, through an antigenic shift the virus becomes significantly different as two viruses can swap segments of their genomes with each other, resulting in a hybrid strain. Because the strain is so different from the preexisting viruses, it is able to escape the immune response, causing outbreaks and pandemics. It is even capable of jumping from one species to another. Such a “shift” occurred in the spring of 2009, when an H1N1 influenza virus with genes from North American Swine, Eurasian Swine, humans and birds emerged to infect people and quickly spread, causing a pandemic.
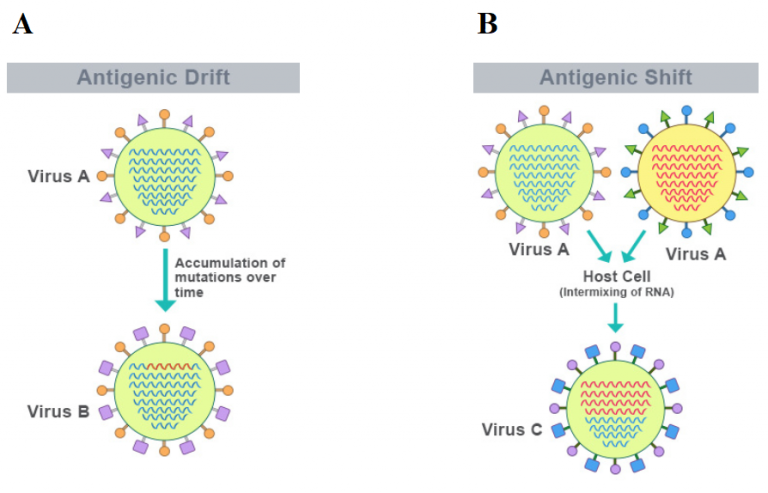
Figure 2: Antigenic drift (A) and antigenic shift (B) process (adapted from https://frontiersmag.wustl.edu/2017/12/15/the-challenges-of-neutralizing-influenza-and-the-quest-for-a-universal-vaccine/).
Influenza does not have the large capacity to modulate our immune system as HCMV does, nevertheless it has adopted various strategies (apart from high mutation rate) to evade the antiviral immune responses of our immune system. The NS1 protein encoded by influenza acts as an antagonist for the host anti-viral responses, as it limits the production and effects of interferons, the primary antiviral agents in the body (17). Besides the direct effect on anti-viral agents, influenza viruses are also able to counteract cells of the innate immune system. For example, influenza virus infection can impair the maturation of dendritic cells and the capacity of the dendritic cells to induce virus-specific T cell responses. Furthermore, it has been shown that influenza virus directly infects, replicates in NK cells and eventually even kills them (18). These strategies are used to secure viral replication and spread to other hosts, all before the immune system of the current host is able to get rid of the infection.
Even though influenza causes a lot of burden on health care, mostly due to the seasonal epidemics, we have gotten quite used to it and accepted it as a state of normal in our lives. When you look back in history and you keep in mind that viruses are constantly around us in great abundance and can move from animal to human, new pandemics are not as unexpected as they might seem. Especially, when a virus has mechanisms to outsmart our immune system and targets our respiratory tract, it easily spreads to other hosts by aerosols in the air and therefore is difficult to contain. Next to the flu, also the current pandemic of the new corona virus COVID-19 has demonstrated that very clearly. Majority of the patients experience mild to moderate symptoms including fever, cough, and general malaise, similar to the flu. Few cases may develop acute respiratory distress syndrome and acute lung injury (19). It is still not fully understood how the virus interacts with our immune system however, it is shown that COVID-19 virus can tone down the activities of the innate immune system by changing some signaling molecules. More importantly, it has been suggested that in some patients the immune system becomes hyperactive, which leads to a so called “cytokine storm”. This can lead to significant damage to vital organs, especially the lungs (20, 21). Surprisingly, COVID-19 seems not to pose a threat to children, but only to elderly and adult patients with pre-existing health conditions, as it is mostly the old and sick, who are not able to turn off their inflammatory response (19, 22). In this regard COVID-19 acts very different from other respiratory diseases like the flu, as these types of viruses affect both the immature immune system and the one that starts to wear out. Another difference so far is that COVID-19 has not mutated much, therefore the chances for an effective vaccine are higher, and risk of new pandemics are lower when compared to the flu (23). But of course, only time will tell.
We have to accept that we will always be challenged by current as well as new viruses. And only a better understanding of the interactions between the virus and the host immune response may lead to new therapeutic and preventative tools. So, who is smarter, us or the virus? Difficult to say. Luckily one thing the past has taught us is that not only the viruses evolve, so do we.
References
1. Arden KE, Mackay IM. Human rhinoviruses: coming in from the cold. Genome Med. 2009;1(4):44.
2. Briese T, Renwick N, Venter M, Jarman RG, Ghosh D, Köndgen S, et al. Global distribution of novel rhinovirus genotype. Emerg Infect Dis. 2008;14(6):944-7.
3. Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, et al. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science. 2009;324(5923):55-9.
4. Wildschutte JH, Williams ZH, Montesion M, Subramanian RP, Kidd JM, Coffin JM. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci U S A. 2016;113(16):E2326-34.
5. Microbiology by numbers. Nature Reviews Microbiology. 2011;9(9):628-.
6. Woolhouse M, Scott F, Hudson Z, Howey R, Chase-Topping M. Human viruses: discovery and emergence. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2012;367(1604):2864-71.
7. Vossen MT, Westerhout EM, Söderberg-Nauclér C, Wiertz EJ. Viral immune evasion: a masterpiece of evolution. Immunogenetics. 2002;54(8):527-42.
8. Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010;20(4):202-13.
9. Rafailidis PI, Mourtzoukou EG, Varbobitis IC, Falagas ME. Severe cytomegalovirus infection in apparently immunocompetent patients: a systematic review. Virol J. 2008;5:47.
10. Manandhar T, Hò GT, Pump WC, Blasczyk R, Bade-Doeding C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. Int J Mol Sci. 2019;20(15).
11. Yang Z, Bjorkman PJ. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U S A. 2008;105(29):10095-100.
12. Nattermann J, Nischalke HD, Hofmeister V, Kupfer B, Ahlenstiel G, Feldmann G, et al. HIV-1 infection leads to increased HLA-E expression resulting in impaired function of natural killer cells. Antivir Ther. 2005;10(1):95-107.
13. Nattermann J, Nischalke HD, Hofmeister V, Ahlenstiel G, Zimmermann H, Leifeld L, et al. The HLA-A2 restricted T cell epitope HCV core 35-44 stabilizes HLA-E expression and inhibits cytolysis mediated by natural killer cells. Am J Pathol. 2005;166(2):443-53.
14. Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci U S A. 2000;97(4):1695-700.
15. Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold ME, et al. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol. 2002;76(3):1285-92.
16. WHO. Influenza (Seasonal): WHO; 2018 [Influenza (Seasonal) fact sheet]. Available from: https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal).
17. Krug RM. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol. 2015;12:1-6.
18. van de Sandt CE, Kreijtz JH, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses. 2012;4(9):1438-76.
19. Worldometer. Updates COVID-19 Coronavirus pandemic: Worldometer; 2020 [COVID-19 Coronavirus pandemic]. Available from: https://www.worldometers.info/coronavirus/.
20. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020.
21. András K, Sükösd H. The complex immunological pathogenesis behind COVID-19. 2020.
22. Molteni M. 2020. Available from: https://www.wired.com/story/kids-can-get-covid-19-they-just-dont-get-that-sick/.
23. Ries L. Healthline2020. Available from: https://www.healthline.com/health-news/what-to-know-about-mutation-and-covid-19.